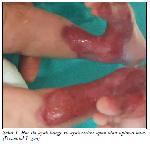
Yirmi dokuz yaşındaki annenin 4. gebeliğinden sezaryen ile miadında 3100 gram doğan erkek bebek her iki ayak bileği ve ayak sırtında cilt defektleri saptanması üzerine AKK tanısı ile yenidoğan servisine yatırıldı (Şekil
1). Annede gebelik döneminde ilaç, sigara ve alkol kullanımı yoktu. Anne - baba arasında 3. derece akrabalık vardı. Diğer üç kardeşi ve anne - baba sağlıklıydı. Fizik incelemede doğum ağırlığı, boy ve baş çevresi normal aralıkta, vital bulguları normal, el ve ayaklarda tırnak deformitesi vardı (Şekil
2). Yüzeyden 1-2 mm çökük olan cilt defektleri normal cilt ile keskin sınırlarla ayrılıyordu. Cilt defekti ince, saydam bir membranla kaplı, zemindeki vasküler yapıları belirgin ve oldukça frajil görünümdeydi. Diğer sistem muayeneleri normal olan hasta dermatoloji bölümüne konsülte edildi.
Büyütmek İçin Tıklayın |
Şekil 1: Her iki ayak bileği ve ayak sırtını içine alan aplazia kutis (Postnatal 1. gün). |
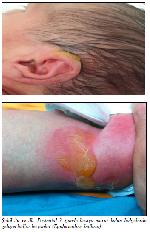
Cilt lezyonlarına steril vazelin, Triticum Vulgare ekstresi içeren krem ve serum fizyolojik ile ıslatılmış steril gazlı bezle konservatif tedavi başlandı. Cilt biyopsisi gerekli görülmedi. Postnatal 3. günde hastanın sırt, kulak kepçesi gibi basıya maruz kalan bölgelerinde büllöz lezyonlar gelişti (Şekil 3a ve 3b).
Büyütmek İçin Tıklayın |
Şekil 3a ve 3b: Postnatal 3. günde basıya maruz kalan bölgelerde gelişen büllöz lezyonlar (Epidermolizis bülloza). |
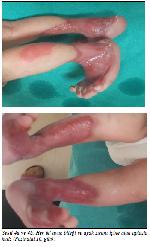
Bu bulgularla hastada Bart sendromu düşünüldü. Aile öyküsü derinleştirildiğinde annenin teyzesinin büllöz cilt lezyonları nedeni ile kaybedildiği ve amcanın benzer bulgularla kaybedilen bebekleri olduğu öğrenildi. Renal ve kraniyal ultrasonografi incelemeleri normaldi. Genetik bölümüne konsülte edilerek anne, baba ve bebekten genetik inceleme için kan ayrıldı. Konservatif tedavinin 10. gününde epitelizasyon oluşmaya başladı (Şekil 4a ve 4b).
Büyütmek İçin Tıklayın |
Şekil 4a ve 4b: Her iki ayak bileği ve ayak sırtını içine alan aplazia kutis (Postnatal 10. gün). |
Nemlendiricili topikal kremlerle aileye yara bakımı için eğitim verildi. Hasta postnatal 30. günde genetik, dermatoloji, plastik ve rekonstriktif cerrahi ve pediatri poliklinik kontrollerine gelmek üzere taburcu edildi. Hasta, takip muayenelere getirilmediği için genetik inceleme yapılamadı. Aile telefonla arandığında hastanın 57 günlükken emmeme, hızlı soluma nedeniyle başka bir merkeze götürüldüğü ve aynı gün öldüğü öğrenildi.


)
)
)
)
)