OLGU 1
Yedi yaşında kız hasta, doğumundan itibaren saçlarının seyrek olması; zor uzaması ve kolay dökülmesi yakınmaları ile kliniğimize başvurdu (Tablo
1). Son bir yıldır kol-diz-bel ağrısı, halsizlik ve saçlarında seyrekleşme-dökülme yakınmasıyla ile akrodermatitis enteropatika tanısı alarak çinko ve B12 vitamini tedavisi yapılmış. Bu tedaviler ile düzelme gözlenmemiş. İki kardeş erken doğum nedeni ile kaybedilmiş olup diğer 3 kardeş sağlıklı olarak saptandı.
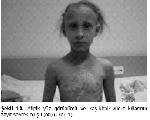
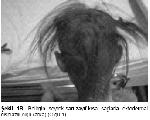
Fizik bakıda deri ince, kuru, hafif buruşuk ve pullanma göstermekte olup sağ kol, diz ve yüzde eski ekimozları vardı. Boyunda 1x1 cm düzensiz kenarlı cafe au lait lekeleri, sırt bölgesinde 3 ve karın derisinde 1 adet 4x1 cm çaplı hipopigmente leke saptandı. Saçlar belirgin kısa olup doğumdan beri hiç kesilmemişti. Kaş, kirpik ve diğer vücut kılları da zayıf-seyrek olan hastada atipik yüz görünümü (çıkık alın, malar hipoplazi, çökük burun kökü, geniş burun delikleri, periorbital hiperpigmentasyon, düsük kulaklar ve yapışık kulak kepçeleri) gözlendi (Şekil 1A, Şekil 1B). Burun içi atrofik ve kurutlu idi.
Büyütmek İçin Tıklayın |
Şekil 1A: Atipik yüz görünümü ve kaş-kirpik-vücut kıllarının zayıf-seyrek olusu (ön) (Olgu 1). |
Büyütmek İçin Tıklayın |
Şekil 1B: Belirgin seyrek-sarı-zayıf-kısa saçlarla ektodermal displazili olgu (arka) (Olgu 1). |
Laboratuarda hemoglobin (Hb): 11.8 g/dl, beyaz küre (WBC): 2.800/mm3 (nötrofil (ANC): 1.680/mm3), trombositler (Plt): 125.000/mm3, eritrosit sedimentasyon hızı (ESR): 23 mm/h olup biyokimyasal değerlerinde patoloji saptanmadı. Serum çinko düzeyi: 68.6 mg/dl (normal: 64-118 mg/dl), serum demiri: 49 mg/dl, total demir bağlama kapasitesi: 270 mg/dl, ferritin: 98 mg/dl ve laktat dehidrogenaz (LDH): 326 U/L idi. Serum immunoglobulin (Ig) G ve A seviyeleri, doğal öldürücü (NK) hücreleri yaşa göre normal oranlarda saptandı. Kanama diyatezi yönüyle yapılan incelemeleri ve kemik iliği aspirasyonu normal olarak değerlendirildi. Gözyaşı bezleri normal olup işitme testleri normaldi.
OLGU 2
Altı yaşında kız hasta ateş, kusma ve şuur bulanıklığı yakınmaları ile başvurdu (Tablo 1). Saç-kasların seyrek olması, az terleme, özellikle gözlerinde yanma/ışığa bakamama şikayetleri vardı. Baş ağrısı, fışkırır tarzda (8-10 kez/gün) kusma yanı sıra uykuya meyil gözlenmiş. Öyküsünden doğumda saç/kaslarının olmadığı, 2 yaşına doğru seyrek çıktığı; çıkan saçlarının yavaş uzadığı ve kolaylıkla döküldüğü öğrenildi. Saçları 6 yaşına kadar yalnızca bir kez kestirilmiş. Annede iki aylıktan itibaren 3 yaşına kadar ateşli nöbet gözlenmiş.
Fizik bakısında şuur konfüze, deri kuru olup yer yer pullanmalar mevcuttu. Karaciğeri 3 cm olarak ele gelen olguda ense sertliği ve babinski refleksi negatif idi.
Laboratuarda Hb: 11 g/dl, WBC: 6.700/mm3 (ANC: 5.600/mm3), Plt: 434.000/mm3 ve ESR: 16 mm/h olup biyokimyasal parametreleri normal idi. Gözde kseroftalmi saptandı. Lomber ponksiyonda hücre görülmedi ve kültürlerinde üreme olmadı. Batın ultrasonografisinde karaciğer boyutu 121 mm olup artmış ve parankim ekosu azalmış (fulminan hepatit açısından anlamlı) saptandı. İntraabdominal diğer organlara ait patoloji izlenmedi. Serum çinko düzeyi: 89 mg/dl olup demir, LDH, Igler ve kemik iliği normal olarak saptandı.
Nişasta testi ile anhidroz (sağlıklı cins ve yaşıtları ile karşılaştırmal) saptandı (Şekil 2A). Deri biyopsisi ektodermal displazi ile uyumlu (yüzeyde hiperkeratoz gösteren epidermis, altındaki dermada sebase glandların tamamen kaybolması; ekrin bezlerin ise oldukça azalması, izlenebilen ekrin bezlerin basıklaşmış ve tek tabakadan ibaret) görüldü (Şekil 2B).
OLGU 3
Üçbuçuk yaşındaki kız hasta yürürken zorlanma, ayak parmaklarının ucuna basarak yürüme, saç ve kaslarının doğumdan itibaren seyrek olması, üst yankesici dişlerinin olmaması yakınmalarıyla başvurdu (Şekil 3A, Tablo 1). Doğumdan itibaren saç ve kaslarının seyrek, diş çıkarmasının ise geç olduğu öğrenildi. Yürümeye 2 yaşında sadece tutunarak ve parmak uçlarına basarak başlamış. Daha önce saçlarının seyrek olması yakınmasıyla doktora götürüldüğü, beslenme bozukluğu ve vitamin eksikliğine bağlı saç kaybı olduğu söylenerek çinko ve çeşitli vitamin preparatları verildiği öğrenildi. Ailede benzer yakınmaları olan başka birey yoktu.
Fizik bakıda vücut ağırlığı 3-10 p, boyu 10-25 p olup cilt hafif kuru, saçları ve kasları seyrekti. Üst iki yan kesici dişi olmayan hastanın dişlerinde çürük ve erken diş kaybı saptanmadı. Sakral bölgede spina bifida okülta saptanan hastanın, nörolojik muayenesinde alt ve üst ekstremitelerde derin tendon reflekslerinde artış ve alt ekstremitelerinde spastik güçsüzlük vardı.
Laboratuar incelemelerinde Hb: 13.1 g/dl, WBC: 7.200/mm3 (ANC:2160/mm3), Plt: 228.000/mm3 olup biyokimyasal paremetreleri normal idi. Kan çinko, folik asit, bakır düzeyleri normal olan hastanın beyin manyetik rezonans görüntüleme (MRI)sinde non-komunike hidrosefali ve korpus kallozum disgenezisi, lumbosakral MRIında sakral düzeyde spina bifida ile uyumlu posterior füzyon defekti saptandı. Nonkomunike hidrosefali açısından takibe alındı.
OLGU 4
Onsekiz aylık kız hasta terlememe, sadece iki adet dişinin olması ve çıkan dişlerinde şekil bozukluğu yakınmaları ile başvurdu (Tablo 1, Şekil 4A, Şekil 4B). Öyküsünden doğduğunda hiç kaşı ve saçı olmayan hastanın cildinin çok ince ve kuru olduğu, hemen hiç terlemesinin olmadığı öğrenildi. İlk dişi 14 aylıkken normale göre daha sivri olarak bozuk şekilli çıkan hastanın özgeçmişinde özellik yoktu. Fizik bakısında ağırlık ve ve boyu 10-25 persentil, baş çevresi 3-10 persentil olup ciltte ve burun mukozasında kuruluk vardı. Hastanın üst damakta biri henüz tam çıkmamış, 2 adet koni şeklinde orta kesici dişi vardı. Tam kan sayımı ve biyokimyasal incelemeleri normal olarak saptandı.


)
)
)
)
)
)
)
)